The storage of electrochemical energy in molecular species is a critical challenge to many energy conversion strategies; from dye-sensitized solar cells to photo/electrocatalytic water splitting to redox flow batteries. Arguably, the best method to store electrochemical energy within molecules is by the formation of chemical bonds coupled to multi-electron oxidation/reduction reactions. For example, water splitting results in the 2e– reduction of 2H+ to H2 to form an H-H bond and the 4e– oxidation of 2H2O to O2 to form an O=O double bond.

A general strategy for achieving multi-electron oxidative chemical bond formation is to force intermediate oxidation states to be unstable withrespect to disproportionation. This is exhibited in the I3–/I– redox couple where 2e– oxidation of I– to I3– is highly favored due to the dispropotionation of an I2.- intermediate (2I2.- –> I– + I3–). A similar situation exists for the O2/H2O redox couple mentioned above. Here, the instability of intermediate H2O2 results in a higher favorability of for the direct 4e– oxidation of H2O to O2.
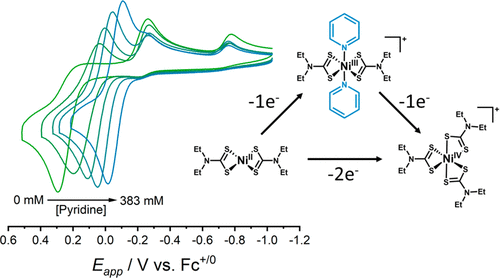
Research in our group targets the development of first-row inorganic coordination compounds that undergo similar redox-cycles. In particular, we are interested in small coordination compounds of nickel and cobalt that undergo changes in coordination environment upon oxidation of Ni(II) or Co(I) (four coordinate) to Ni(IV) or Co(III) (six coordinate). By fine tuning the association constants for metal-ligand bond formation, we can to force the disproportionation of Ni(III) or Co(II) intermediates and thus generate a strongly favored 2e– redox couple that is controlled by metal-ligand bond formation. With this chemistry we seek to ask fundamental questions about multi-electron transfer reactions.
Publications related to this work:
- “Nickel-Based Two-Electron Redox Shuttle for Dye-Sensitized Solar Cells in Low Light Applications” ACS Appl. Energy Mater. 2024, 7, 3645-3655.
- “Zinc-Catalyzed Two-Electron Nickel(IV/II) Redox Couple for Multi-Electron Storage in Redox Flow Batteries” Inorg. Chem. 2022, 61, 19039-19048.
- “Controlling One-Electron vs Two-Electron Pathways in the Multi-Electron Redox Cycle of Nickel Diehtyldithiocarbamate” Inorg. Chem. 2021, 60, 13388-13399.
- “Influence of Pyridine on the Multielectron Redox Cycle of Nickel Diethyldithiocarbamate” Inorg. Chem. 2019, 58, 15371-15384.
